- Home
- Resources
- Newsletters
- Understanding the...
Understanding the EU Pharma Package and how to prepare for its implementation in 2028
Posted on March 19, 2026


After the original proposal from the European Commission in 2023 to review the current EU pharmaceutical incentives and after several years of negotiations between the European Commission, Parliament and Council, the European Union is to implement the most significant reform of its pharmaceutical regulatory framework in more than two decades. In December 2025, provisional agreement was reached on the new “EU Pharma Package”, a comprehensive overhaul that will reshape how medicines are developed, authorised, protected, and commercialised across the EU. The final texts of the new EC Directive and Regulation have now been published.
The new Legislation, which is to take effect in in 2028, aims to address better patient access to medicines, increase innovation in areas of unmet need, including antimicrobial resistance (AMR), and to reduce shortages of medicine. In particular, the Directive focuses on adapting the current exclusivity periods more towards therapeutic products addressing unmet needs (the calculations of which will be more complex) incentivising launch of new antibiotic products and focussing on facilitating drug supply in EU member states.
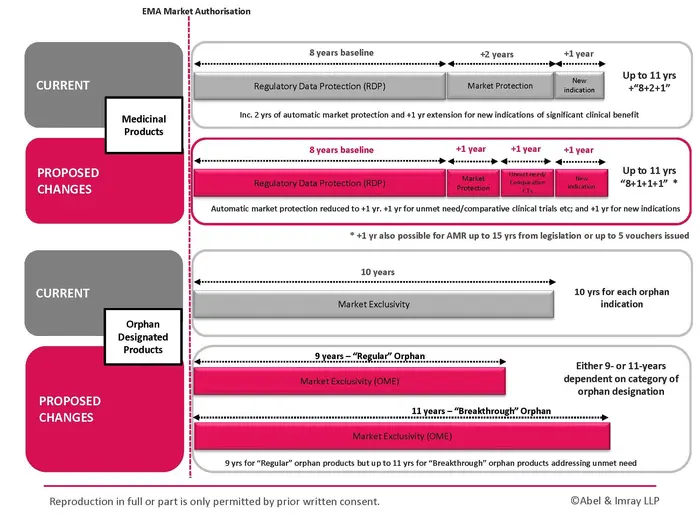
You can view our 'EU Pharma Package Overview Chart' at the end of this article or download it using the Download button on the right-hand side.
What is the EU Pharma Package?
The EU Pharma Package will consist of a new EU Regulation and a new EU Directive that will replace several core Legal Instruments governing medicinal products, including the current Directive 2001/83/EC and Directive 2009/35/EC Regulation (EC) No. 726/2004, the Orphan Regulation (EC) No. 141/2000 and the Paediatric Regulation (EC) No. 1901/2006. Regulation (EC) No. 1394/2007 and Regulation (EU) No. 536/2014 will be amended. In short, it means major changes for the Regulatory Exclusivity (Data and Market) protection of pharmaceuticals, in particular. Below is an overview of the key changes.
Regulatory Exclusivity (RDP)
One of the most consequential aspects of the EU Pharma Package is the complete re-design of the Regulatory Exclusivity framework, replacing the long‑established “8+2(+1)” year term for Regulatory Data and Market Protection (RDP).
Under current rules, new medicinal products benefit from 8 years of Regulatory Data Exclusivity. During this period, a generic or biosimilar company cannot rely on the originator’s clinical and pre‑clinical data to obtain Regulatory Marketing Authorisation for its generic or biosimilar product. This 8 years of baseline data protection remains the same.
However, the two-year (+2) period of Regulatory Market Exclusivity, which currently automatically follows the Regulatory Data Exclusivity period, will be reduced to a one-year (+1) period under the new legislation. During this period, generics and biosimilars can make preparations for approval of their products relying on the Originator’s data to demonstrate safety and efficacy but they cannot launch their products until the end of this Market Exclusivity period.
It is currently possible to obtain an additional one-year (+1) period of Market Exclusivity if the authorisation holder obtains an authorisation for an indication of “Significant Clinical Benefit”, however, under the new legislation, it will be possible to extend the 8-year period of Regulatory Data Exclusivity and (+1) year of Regulatory Market Exclusivity by two separate (+1)(+1) additional year periods for the following conditions, up to a maximum combined Exclusivity period of 11 years:
- addressing an unmet medical need; or
- meeting certain conditions, such as comparative clinical trials; or
- carrying out clinical trials in more than one EU member state under certain conditions (+1 year); and
- adding new indications that offer “Significant clinical benefit” (+1 year).
These changes are aimed at incentivising research and development into rare and ultra-rare diseases, and to reward research into products that address an unmet or a high unmet clinical need. Such exclusivity protection periods will be shorter, more complex and more nuanced for the majority of molecules creating some uncertainty around how much Regulatory Market Exclusivity may be obtained. It is also likely that more data, clinical trials in more than one EU member state or comparative data will be necessary to achieve the maximum protection available.
Re-purposed Products
Re-purposed medicinal products are known active substances that obtain market authorisation for a new therapeutic indication wherein a significant clinical benefit has been shown.
Under Article 84 of the new Directive a 4-year period of Regulatory Data Protection will be granted to such products provided that:
- adequate clinical studies and, where relevant, non-clinical studies/tests are carried out in relation to the therapeutic indication demonstrating that it is of Significant Clinical benefit; and
- the medicinal product is authorised and has not previously benefitted from data protection, or 25 years have passed since the granting of the initial marketing authorisation of the medicinal product concerned.
It should be noted that the Regulatory Data Protection may only be granted once for any given medicinal product and that it will be necessary to indicate on the Marketing Authorisation (MA) that the medicinal product authorised in the EU has been authorised with an additional therapeutic indication.
Orphan Medicines
For an Orphan designation, the current framework provides for 10 years of Regulatory Market Exclusivity per Orphan indication per Orphan medicinal product. This period may currently be extended by 2 years where a Paediatric Investigation Plan (PIP) has been completed. This period exists in parallel with RDP.
The protection for “Regular” Orphan medicinal products under the new legislation will be reduced to 9 years of Orphan Market Exclusivity (OME).
However, under the new legislation, a “Breakthrough” category of Orphan medicinal products will be introduced for medicines addressing high unmet need. This category includes medicinal products for diseases with no available EU treatment where strong evidence of clinical benefit is shown. Under the new legislation, the protection for Breakthrough products will be 11 years of OME. To be awarded the extended OME, an Orphan MA holder will have to show evidence of clinically relevant reduction in disease morbidity or mortality for the relevant patient population.
4 years of OME will be available for Orphan Medicinal Products authorised based on bibliographical data. An application based on Article 13 of the new Directive and Article 71 of the new Regulation may only be submitted if the applicant can demonstrate that: (a) no reference medicinal product is or has been authorised in the EU for the active substance of the medicinal product at the time of submission of the Market Authorisation application; or (b) whilst a reference medicinal product for the active substance of the medicinal product concerned has been authorised, it is not available on the market within the EU.
The proposal of a new “Global Orphan Marketing Authorisation” Concept has now become part of the new Legislation. Where there is more than one therapeutic indication, the protection for an Orphan medicinal product will be considerably reduced under the new legislation. The intention of the new legislation is to prevent “evergreening” of the same active ingredient where an MA holder holds more than one MA for the same active substance. It will only be possible to extend Orphan Market Exclusivity by a one-year (+1) period for each new Orphan therapeutic indication to a maximum of (+2) years rather than the current situation of 10 years of OME per indication running in parallel. (This Concept works in a similar way to the Global Market Authorisation for Regulatory Data Exclusivity which has been in place for a number of years).
An additional feature of the new legislation, which also effectively reduces the OME, is that the submission, validation and assessment of similar medicinal products during the last two years before the OME expires is now allowed. Such similar products may be placed on the market immediately after expiry of the OME.
Paediatric Exclusivity
The reward of 6-month extension to a Supplementary Protection Certificate (SPC) will remain, following completion of a PIP linked to the first approved indication.
It is also currently possible to obtain an additional year (+1) of Market Exclusivity for a new indication of “Significant Clinical Benefit” under RDP with paediatric conditions being one type of condition which could be considered to fall under the definition of “Significant Clinical Benefit”.
It should be noted that there can be no double rewards for the same paediatric indication i.e. one must choose an extension to the SPC or to the RDP. This provision remains the same as previously.
The current 2-year (+2) reward for a paediatric indication with regard to Orphan Products will, however, no longer be available under the new proposals. As such, the protection for an Orphan medicinal product will be reduced under the new legislation.
Anti-microbial Resistance (AMR)
There is a new incentive for launching antimicrobial products under the legislation. This takes the form of a “Transferable Data Exclusivity Voucher” for priority antimicrobial products. This will give an additional year (+1) of Regulatory Data Exclusivity for one authorised product. The additional year of data protection may be used once, for the priority antimicrobial or for another centrally authorised medicinal product of the same or a different MA holder.
However, there are certain conditions that apply in order to obtain a voucher including a limitation on gross sales. In addition, a voucher may only be obtained in the first 15 years after entry into force of the new legislation, or until 5 vouchers have been issued, whichever occurs first.
Expanded Bolar Exemption
The Bolar exemption is a legal provision that allows clinical trials and other launch preparations for generic and biosimilar medicines to be carried out before the expiry of a patent or an SPC, without this being considered to be patent infringement.
To date, Article 85 of the EU Directive 2001/83/EC has only covered clinical trials and studies strictly necessary for marketing authorisation, although it has been implemented more broadly when adopted into the national laws in numerous European countries to also include clinical trials and other launch preparations for innovators.
The EU Pharma Package aims to improve access to affordable medicines and reduce legal uncertainties by harmonisation and clarification of Article 85 of the new Directive.
These reforms will make it easier for generic and biosimilar manufacturers to be ‘launch-ready’ once the patent or the SPC expires.
The main changes will benefit both generic and biosimilar companies as follows: -
- Clearer Definitions: Article 85 of the EU Directive provides explicit guidance on permissible activities under the exemption, including those necessary for approval processes, health technology assessments (HTAs) and pricing procedures;
- Expanded Scope: The revised rules broaden the exemption to cover activities such as manufacturing, storage and import by 3rd party providers, which were previously ambiguous; and
- Harmonization: By addressing inconsistencies in national implementations (caused by different EU countries having different wording for the Bolar exemption written into their law), the proposal seeks to reduce legal uncertainty and streamline processes across the EU.
What’s next?
The final approval of the published text of the new EC Directive and Regulation by the EU Parliament and Council will occur in summer 2026. A transition phase to allow time for EU Member States to update their national laws to reflect the new rules will be given between 2026 and 2028. The new pharmaceutical legislation will be formally adopted in 2028.
The recently published changes will allow quicker and more coordinated generic and biosimilar entry into the market, with reduced periods of protection in terms of RDP and Orphan Market Exclusivity for innovators. In respect of Orphan medicinal products previously relying on separate terms of Orphan Market Exclusivity for each therapeutic indication, it will be necessary to plan to obtain Orphan Market Authorisations for further therapeutic indications within 2 years before the end of the Orphan Market Exclusivity period of the first authorised indication. In addition, the expansion of the Bolar exemption allows for generics and biosimilars to hit the market the moment patent protection expires.
Extensions of the baseline Regulatory Exclusivity periods will be available but are likely to be more difficult to achieve in practice. We are also likely to see much more uncertainty regarding whether an extension of Regulatory Market Exclusivity may be granted, given the increased need to demonstrate clinical effectiveness, carry out comparative clinical trials or meet an unmet need. Such protection is less important where primary protection for a product is provided by one or more patents which outlast the Regulatory Data Exclusivity, but it will be very important where it is intended to rely on other types of exclusivities as the primary protection for a product.
Overall, innovators will need stronger and more robust patent portfolios, given that biosimilar and generic products may be on the market earlier than was previously possible. More careful planning will be necessary, to retain their competitive edge against increasingly well-prepared generic competitors.
Our team will be offering a package to assist organisations with reviewing their patent portfolio in order to assess the impact of these changes and to provide you with tailored advice. Many of our attorneys have worked in‑house within the pharmaceutical industry, so we can provide practical, experience‑led advice tailored to your needs.
For more information, and if you would like guidance on strengthening your IP position, navigating the new framework, or assessing how these reforms affect your portfolio, please contact Lindsey Kent, Chris Lindsay, or your usual Abel + Imray advisor.